|
|

|

|
| e-Pub |
Section: New Results
Integrative RNA structural modeling
To circumvent expensive, low-throughput, 3D experimental techniques such as X-ray crystallography, a low resolution/high throughput technology called SHAPE is increasingly favored for structural modeling by structural biologists.
|
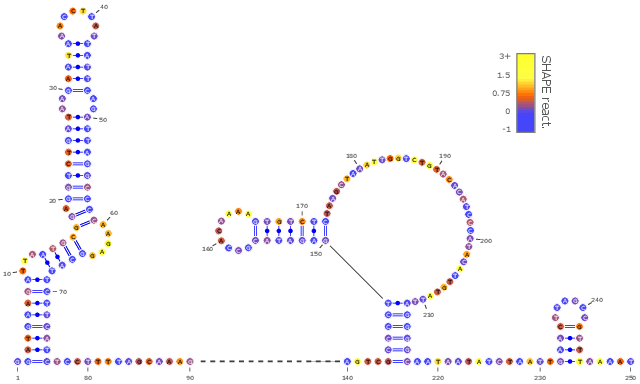
Within Afaf Saaidi's thesis, funded by the Fondation pour la Recherche Médicale and co-supervised by Bruno Sargueil at Faculté de Pharmacie of Université Paris V, we have developed integrative modeling strategies based on Boltzmann sampling. Preliminary results, obtained by applying these methods to model the structures of 3'UTR regions in Ebola, were presented at JOBIM 2016 [14].
Moreover, in collaboration with McGill University (Canada), we cross-examined mutate-and-map data (MaM [30]) in the light of evolutionary data. MaM data consist in the sequential SHAPE probing of a set of mutant RNAs, obtained by systematic point-wise mutations, to highlight structurally-dependent nucleotides, later to use dependent pairs as constraints in (an automated) structural modeling. We chose to adopt an alternative perspective on MaM data, and used the perturbation of the SHAPE profiles as a proxy for the structural disruption induced by a mutation. Disruptive mutations are rescued within homologs, i.e. compensated to re-establish the structure. However, our analysis also revealed the existence of non-structurally local (neither on the 2D or 3D levels) nucleotides which have significant mutual-information with highly disruptive positions, despite not being involved in any obvious compensatory relationship.
We hypothesized that such mutations are revealing of interactions involving RNA. In a manuscript published in Nucleic Acids journal, we tested and validated this hypothesis by showing its capacity to discriminate discriminative positions that are known to be in contact with specific ligands (proteins, DNA, small molecules...) [10].
|
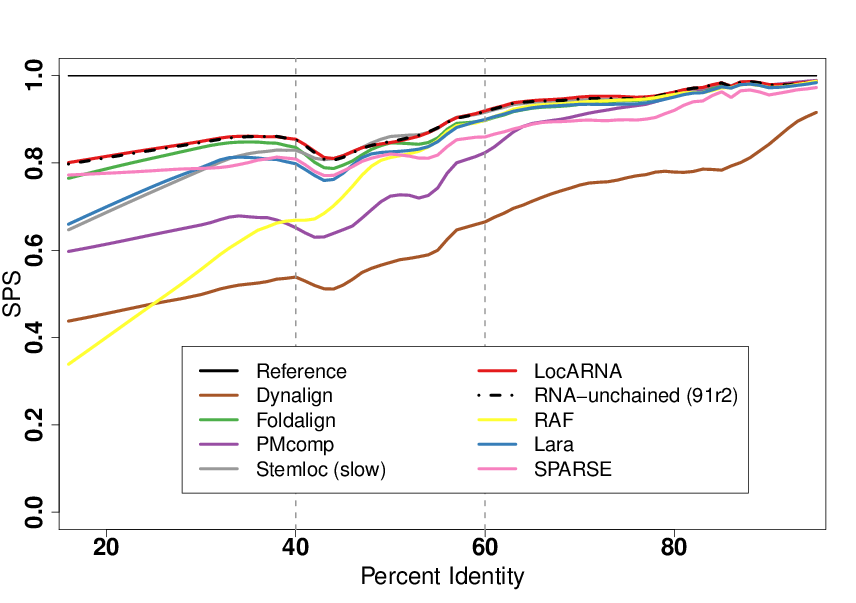
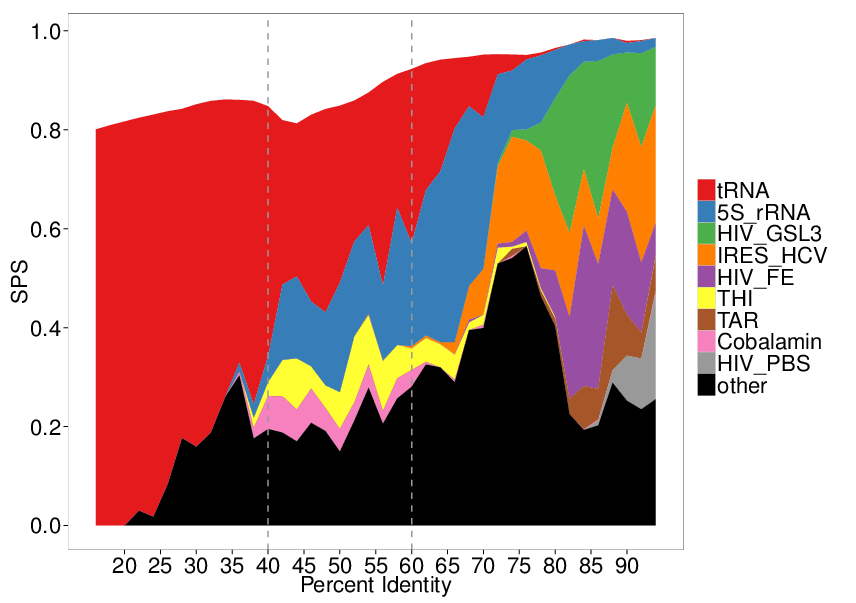
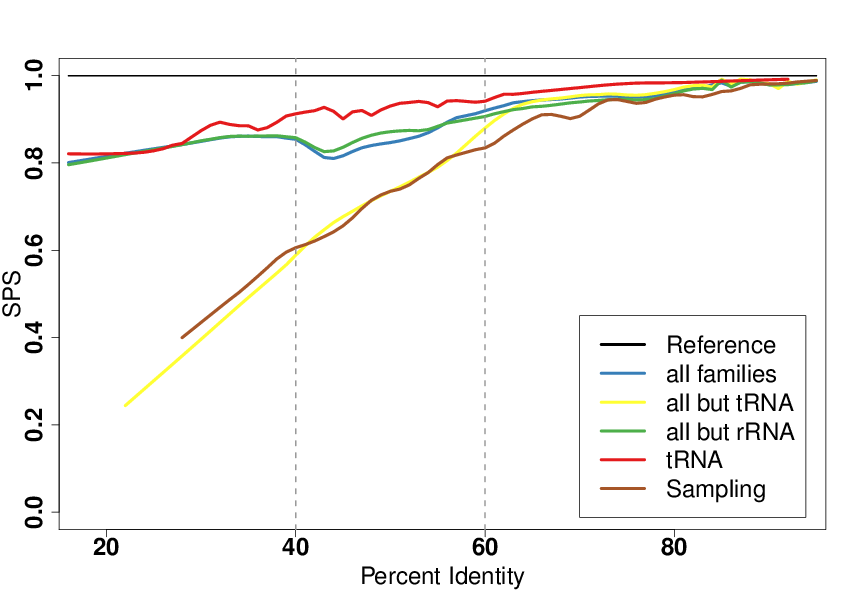
A fruitful line of research for RNA structure prediction is based on a comparative approach. Whenever homologous RNAs are identified, a classic strategy is to perform a simultaneous alignment and folding of several RNAs. Many software (30+) have been contributed over the past decades for this problem, leading to the introduction of benchmarks, one of the most prominent being the Bralibase , to position new developments and identify axes of progression. One such desired improvement, as illustrated in Figure 5, was the difficulties experienced by most software around the 40-60% sequence identity range, which was believed to arise from deep algorithmic reasons. In collaboration with Cedric Chauve (Simon Fraser University, Canada) Benedikt Löwes and Robert Giegerich (Bielefeld University, Germany), we showed that this perceived difficulty was simply the consequence of a strong bias towards tRNAs in the 40-60% sequence identity region. Moreover, we argued that the overall performance of existing tools for low sequence identities were largely overestimated [8].
Finally, we presented at JOBIM 2016 an efficient implementation, called LicoRNA , of our parameterized complexity algorithm based on tree-decomposition for the sequence/structure alignment of RNA [15]. Specifically, we showed that our LicoRNA , by including an expressive scoring scheme and capturing pseudoknots of arbitrary complexity, generally outperforms previously contributions for the problem.